Aneuploidias dos cromossomos sexuais
Síndrome de Klinefelter
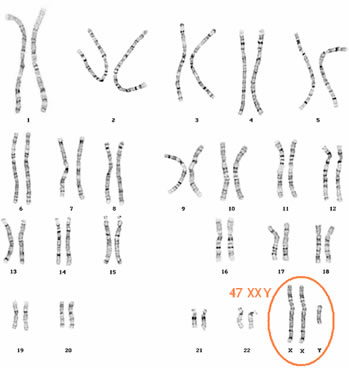
São indivíduos do sexo masculino que apresentam cromatina sexual e cariótipo geralmente 47 XXY.
Eles constituem um dentre 700 a 800 recém-nascidos do sexo masculino, tratando-se portanto de uma das condições intersexuais mais comuns. Outros cariótipos menos comuns são 48 XXYY; 48 XXXY; 49 XXXYY e 49 XXXXY que, respectivamente, exibem 1, 2. e 3 corpúsculos de Barr.
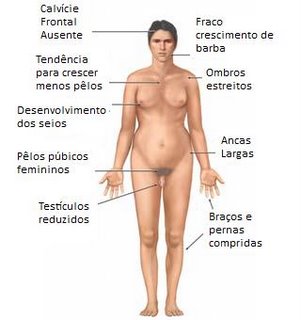
Embora possam ter ereção e ejaculação, são estéreis, pois seus testículos são pequenos e não produzem espermatozoides devido à atrofia dos canais seminíferos. Outras características muitas vezes presentes são: estatura elevada, corpo eunucoide, pênis pequeno, pouca pilosidade no púbis e ginecomastia (crescimento das mamas).
Além dessas alterações do sexo fenotípico, os pacientes com Síndrome de Klinefelter apresentam uma evidente diminuição do nível intelectual, sendo esta tanto mais profunda quanto maior for o grau da polissomia.
Ao contrário do que ocorre na Síndrome de Turner, os pacientes Klinefelter apresentam problemas no desenvolvimento da personalidade, que é imatura e dependente, provavelmente em decorrência de sua inteligência verbal diminuída.
Até 1960, a prova definitiva para o diagnóstico era fornecida pelo exame histológico dos testículos que, mesmo após a puberdade, revela ausência de células germinativas nos canais seminíferos; raros são os casos de Klinefelter férteis que, evidentemente, apresentam alguns espermatozoides normais. Atualmente a Identificação dos Klinefelter é assegurada pelo cariótipo e pela pesquisa da cromatina sexual.
Sindrome do triplo X ou Super fêmea
Mulheres com cariótipo 47 XXX ocorrem numa frequência relativamente alta: 1 caso em 700 nascimentos aproximadamente. Elas apresentam fenótipo normal, são férteis, mas muitas possuem um leve retardamento mental. Apresentam corpúsculo de Barr.
Os casos de mulheres 48 XXXX e 49 XXXXX são raros e se caracterizam por graus crescentes de retardamento mental.
Sindrome do duplo Y ou Super macho
Indivíduos com cariótipo 47,XYY ocorrem com a frequência de 1 caso por 1.000 nascimentos masculinos.
Embora sejam, na maioria, homens normais, os primeiros estudos sugeriam que entre eles ocorria uma frequência extremamente alta de pacientes retardados mentalmente e com antecedentes criminais; tais estudos revelaram que cerca de 2% dos pacientes Internados em instituições penais e hospícios tinha este cariótipo, o que mostrava serem os indivíduos XYY internados 20 vezes mais numerosos (em lugar de 1 por mil, 2% corresponde a 20 por mil) do que na população livre.
No entanto, os mesmos dados revelaram que 96% dos indivíduos XYY são normais. Deste modo, tornam-se necessárias pesquisas mais amplas antes de se relacionar essa constituição cromossômica particular com determinados traços anormais de comportamento; é especialmente importante evitar uma interpretação Ingênua relacionada com um “cromossomo do crime”.
Uma característica física bem evidente dos XYY é a estatura elevada, pois eles geralmente têm mais de 180 cm, ou seja. são 15cm mais altos do que a média dos indivíduos masculinos cromossomicamente normais.
Podemos sugerir que genes localizados no cromossomo Y elevam a estatura e predispõem seus portadores para comportamentos inesperados; de fato, o perfil psicológico do indivíduo XYY inclui imaturidade no desenvolvimento emocional e menor inteligência verbal, fatos que podem dificultar seu relacionamento interpessoal. Um fato digno de nota é que os pacientes institucionalizados, tanto XY como XYY, exibem uma taxa de testosterona aumentada, o que pode ser um fator contribuinte para a inclinação anti-social e aumento de agressividade.
Síndrome de Turner (XO)
É uma monossomia na qual os indivíduos afetados exibem sexo feminino mas geralmente não possuem cromatina sexual.
O exame de seu cariótipo revela comumente 45 cromossomos, sendo que do par dos cromossomos sexuais há apenas um X; dizemos que esses indivíduos são XO (xis-zero), sendo seu cariótipo representado por 45 X. Muitas dessas concepções terminam em aborto; é provável que 97% desses conceitos sejam eliminados chegando a termo apenas 3%, de modo que essa monossomia constitui uma das causas mais comuns de morte Intra-uterina. Por isso é uma anomalia cromossômica rara, atingindo apenas 1 entre 3000 mulheres normais.
Trata-se, fundamentalmente, de mulheres com disgenesia gonadal, isto é, cujos ovários são atrofiados e desprovidos de folículos; portanto, essas mulheres não procriam, exceto em poucos casos relatados de Turner férteis, em cujos ovários certamente há alguns folículos.
Devido à deficiência de estrógenos elas não desenvolvem as características sexuais secundárias ao atingir a puberdade, sendo, portanto, identificadas facilmente pela falta desses caracteres; assim, por exemplo, elas não menstruam (isto é, têm amenorreia primária). Quando adultas apresentam geralmente baixa estatura, não mais que 150 cm; infantilismo genital – clitóris pequeno, grandes lábios despigmentados, escassez de pêlos pubianos; pelve androide, isto é, masculinizada; pele frouxa devido à escassez de tecidos subcutâneos, o que lhe dá aparência senil; unhas estreitas; tórax largo e em forma de barril; alterações cardíacas e ósseas. No recém-nascido frequentemente há edemas nas mãos e nos pés, o que leva a suspeitar da anomalia.
As primeiras observações realizadas com indivíduos severamente afetados associavam a síndrome de Turner algum grau de deficiência mental. Posteriormente ficou evidente que estas pacientes têm um desenvolvimento cognitivo alterado apenas qualitativamente, pois elas possuem uma inteligência verbal superior à das mulheres normais, compensando, assim, as suas deficiências quanto à percepção forma-espaço. Disto resulta que o nível intelectual global das Turner é igual ou, mesmo, levemente superior ao da população feminina normal.
Por outro lado, não exibem desvios de personalidade, o que significa, inclusive, que sua identificação psicossexual não é afetada. Em decorrência da disgenesia ovariana, a única fonte de estrógenos para essas pessoas são as supra-renais; como a taxa desses hormônios é baixa, as pacientes devem receber aplicações de estrógenos para estimular o desenvolvimento dos caracteres sexuais secundários e o aparecimento da menstruação. Usualmente esse tratamento tem início aos 16 anos para evitar que os estrógenos aplicados retardem ainda mais o crescimento.
Hermafroditismo
Por análise do cariótipo é sabido que não se trata de uma síndrome genética (mono ou trissomia halossômica), relacionada aos cromossomos sexuais X ou Y. No entanto, pode estar associado a uma ocorrência de dispermia, havendo fecundação normal (espermatozoide e ovócito de segunda ordem - óvulo) e outra fecundação paralela anômola (espermatozoide e um glóbulo polar – óvulo não diferenciado, em tese, inativo).
Naturalmente, os indivíduos portadores dessa anomalia somente revelam o hermafroditismo durante a puberdade, desencadeando transtornos psicossociais quando descoberto.
Dependendo do tipo anatômico aparente, o período de amadurecimento corpóreo, pode devido a estímulos hormonais, iniciar: o processo menstrual, bem como a ginecomastia (crescimento das mamas) em indivíduos criados como se fossem homens; e falha menstrual, crescimento do clitóris e surgimento de pêlos nos indivíduos criados como se fossem mulheres.



Comentários
Postar um comentário